Enfermedad de Von Hippel Lindau
La Enfermedad de Von Hippel Lindau es una enfermedad rara, hereditaria y multisistémica, caracterizada por el desarrollo, en edades tempranas, de tumores en diferentes localizaciones: oculares, renales, de glándulas suprarrenales, páncreas y sistema nervioso central (sistema formado por el encéfalo y la médula espinal).
Fue descrita por primera vez, en 1895, por Eugene von Hippel, quien describió, que los angiomas (tumor caracterizado por la hiperplasia, desarrollo excesivo de los tejidos, del tejido vascular sanguíneo) de retina eran hereditarios y posteriormente Arvin Lindau, en 1926, sospechó que estos tumores formaban parte de una lesión más amplia del sistema nervioso central.
Durante las primeras décadas del siglo XX se describieron familias cuyos afectados no sólo presentaban angiomas en la retina o el sistema nervioso central sino, además, quistes (saco cerrado debajo de la piel que puede contener un contenido líquido o semisólido) y tumores de órganos viscerales, incluyendo riñones, glándulas adrenales, páncreas y epidídimo (conjunto de vasos seminales situados por encima del testículo).
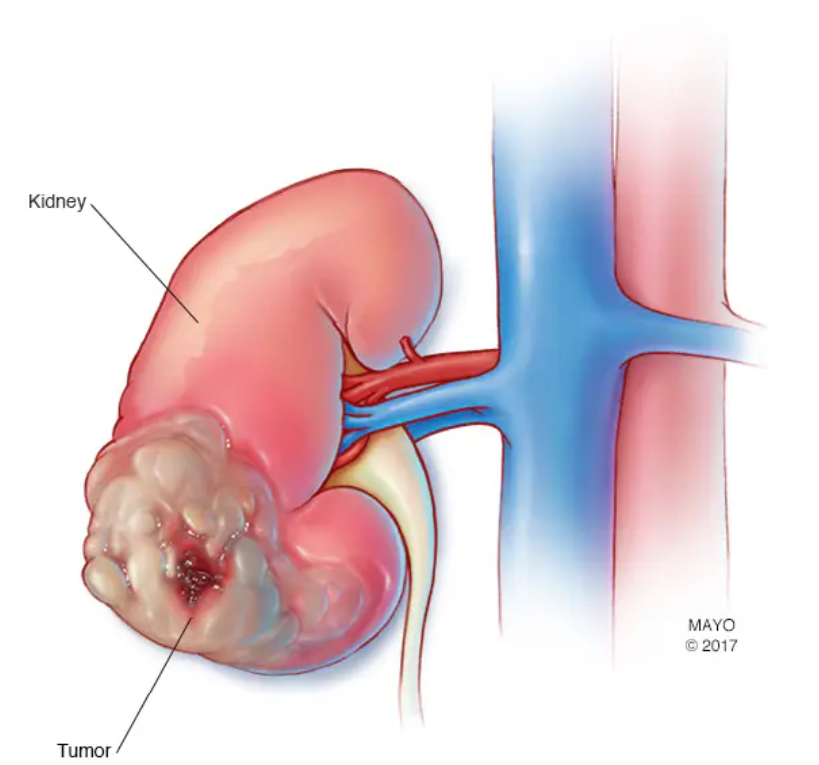
Estos tumores incluyen específicamente carcinoma renal de células claras, cistoadenomas pancreáticos, tumores en los islotes pancreáticos y feocromocitomas.
En 1964 Melmon y Rosen acuñaron el término Von Hippel Lindau y propusieron una definición diagnóstica de esta enfermedad que es la siguiente: un hemangioblastoma asociado con un tumor visceral típico, o un hemangioblastoma o tumor característico vinculado con una historia familiar de Von Hippel Lindau.
Se estima una prevalencia (número de casos de una enfermedad en una población) de 1/35.000 habitantes. Casi todos los casos se presentan antes de los 65 años, aunque la enfermedad suele diagnosticarse a una edad promedio de 26 años.
La Enfermedad de Von Hippel Lindau se debe a un defecto genético. El gen Von Hippel Lindau normal actúa como “gen tumor supresor”, cuya función es suprimir la formación de tumores. Para que se forme un tumor, ambas copias del gen, una del padre y una de la madre, deben inactivarse.
En un individuo que no padece la alteración hereditaria en el gen Von Hippel Lindau, es necesario que ambas copias muten para que se forme un tumor, debido a la desactivación del gen Von Hippel Lindau.
En el caso de personas que heredan una copia defectuosa del gen, sólo basta desactivar la copia restante para que se forme el tumor. Por ello los tumores se forman antes y en más órganos que en las personas normales, ya que todas las células del organismo del individuo tienen una copia alterada del gen desde el nacimiento.
Los afectados presentan uno o varios tumores característicos, y/o una historia familiar concomitante.
Aunque los miembros de un grupo familiar pueden desarrollar un patrón similar de tumores, la expresión de la enfermedad es característicamente variable en cada caso.
Así, los parientes en primer grado pueden desarrollar tumores en diferentes órganos, en número y agresividad diversa y con complicaciones diferentes para tumores por otra parte similares.
Pocos pacientes desarrollan la gama completa de manifestaciones posibles y cerca del 50% de los afectados presenta sólo una manifestación de Von Hippel Lindau.
Numerosos afectados no desarrollan expresiones de la enfermedad hasta una edad avanzada.
La manifestación inicial más frecuente es la angiomatosis en la retina y los hemangioblastomas en el cerebelo, apareciendo posteriormente tumores en cerebro, médula espinal, feocromocitoma en las glándulas suprarrenales, cistoadenoma seroso microquístico en el páncreas y carcinomas de células renales.
También han sido descritas lesiones angiomatosas en hígado, riñón, páncreas, pulmón, piel y epidídimo.
La Enfermedad de Von Hippel Lindau se clasifica en dos tipos en función de la presencia o ausencia de feocromocitomas
- Tipo 1: aquellos que no presentan feocromocitomas, suponen alrededor del 80% de los casos.
- Tipo 2: con presencia de feocromocitomas, aproximadamente un 20% de los casos, de peor pronóstico que los de tipo 1.
- Tipo 2A: caracterizado por la ausencia de carcinomas de células renales y quistes pancreáticos.
- Tipo 2B: con presencia de carcinomas de células renales y quistes pancreáticos, que constituye el grupo con mayor mortalidad y peor pronostico.
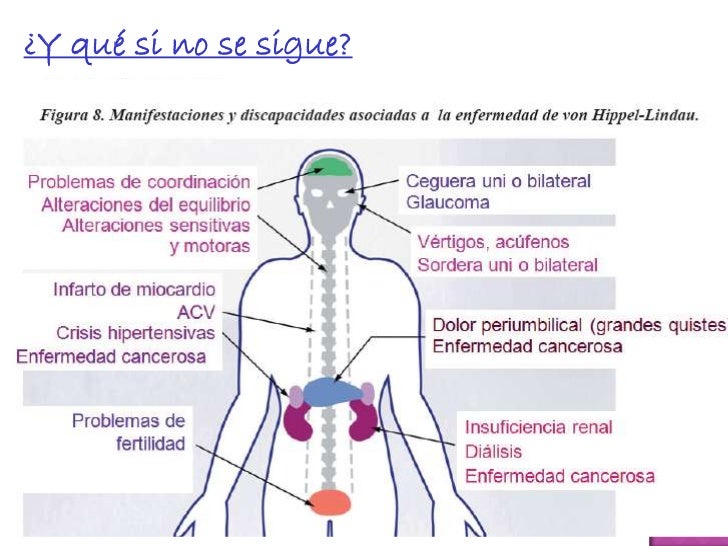
La enfermedad de Von Hippel Lindau presenta una gran variabilidad clínica y los síntomas dependen del tamaño y de la localización de los tumores. Así: Las angiomatosis en la retina pueden causar desprendimiento de retina hemorragias y eventualmente ceguera. Los hemangioblastomas del sistema nervioso central producen síntomas diversos como: cefalea (dolores de cabeza), inestabilidad de la marcha, vómitos, alteraciones del equilibrio y debilidad en extremidades superiores e inferiores.
Los feocromocitomas pueden ser asintomáticos o producir una gran variedad de síntomas, siendo los más frecuentes: jaquecas, sudoración, palpitaciones con o sin taquicardia (latido anormalmente rápido del corazón), nerviosismo, pérdida de peso, dolores abdominales y torácicos, náuseas, vómitos, astenia (debilitación del estado general), hipertensión arterial, hipotensión ortostática (disminución notable de la presión arterial en la posición vertical, que puede acompañarse de vértigo y lipotimia), hiperhidrosis (sudoración excesiva) y arritmias (latido irregular del corazón).
Otros síntomas menos comunes son: alteraciones visuales, disnea (dificultad en la respiración), parestesias (sensación anormal de los sentidos o de la sensibilidad), poliuria (secreción y emisión extremadamente abundante de orina), polidipsia (sed excesiva), mareos, crisis tipo gran mal (variedad de epilepsia caracterizada por crisis que atacan de golpe a todo el cuerpo, con pérdida de conocimiento, caída y trastornos motores, generalmente contracturas y sacudidas rítmicas) aparente, palidez, bradicardia (latido cardíaco inusualmente lento), hematuria (presencia de sangre en la orina) indolora, disartria (dificultad para articular palabras) y temblor.
La enfermedad presenta un amplio espectro de severidad clínica, siendo el carcinoma renal de células claras y el hemangioblastoma de cerebelo, médula espinal o tronco del encéfalo las causas de muerte en la mayoría de los casos.
El diagnóstico de sospecha de la Enfermedad de Von Hippel Lindau se basa en la clínica y los antecedentes familiares y se confirma mediante estudio molecular.
La gran variedad de síntomas en los afectados dificulta el diagnóstico y cada manifestación posible de la enfermedad se detecta de manera diferente.
Si el historial familiar del paciente muestra casos que permitan suponer la presencia de Von Hippel Lindau es importante iniciar los exámenes antes de la aparición de síntomas.
Se recomienda que se inicien los exámenes en niños no mayores de 6 años, empleando técnicas indoloras que no requieran radiación ni inyección de contrastes.
No deben faltar exploraciones oculares y físicas, con especial atención a la tensión arterial y a la evaluación neurológica.
Entre los 10 y 12 años se iniciarán exploraciones cerebrales, mediante escáner y resonancia magnética nuclear, estudios ecográficos de abdomen y determinaciones analíticas de laboratorio.
Numerosos casos demuestran que los afectados suelen vivir más tiempo y con mejor calidad de vida si mantienen una vigilancia constante y se someten a los exámenes con regularidad.
El diagnostico diferencial hay que realizarlo fundamentalmente con la neoplasia endocrina múltiple en la que se presentan feocromocitomas acompañados de diversos tumores, pero la historia familiar de estos pacientes muestra invariablemente la presencia de cáncer medular de tiroides, lo que no ocurre con los pacientes de Von Hippel Lindau.
Actualmente se considera que el tratamiento más eficaz es la prevención de complicaciones relacionadas con crecimientos tumorales. Este enfoque precisa de un diagnóstico presintomático y un seguimiento periódico a lo largo de la vida, realizado por un equipo multidisciplinario. El tratamiento puede incluir:
- 1 – Información genética.
- 2 – Terapia con láser de los angiomas de retina.
- 3 – Resección quirúrgica de hemangioblastomas en el sistema nervioso central
- 4 – Resección de tumores sólidos y quistes renales.
- 5 – Seguimiento médico y resección quirúrgica de feocromocitomas, tumores de los islotes pancreáticos o adenocarcinomas del páncreas.
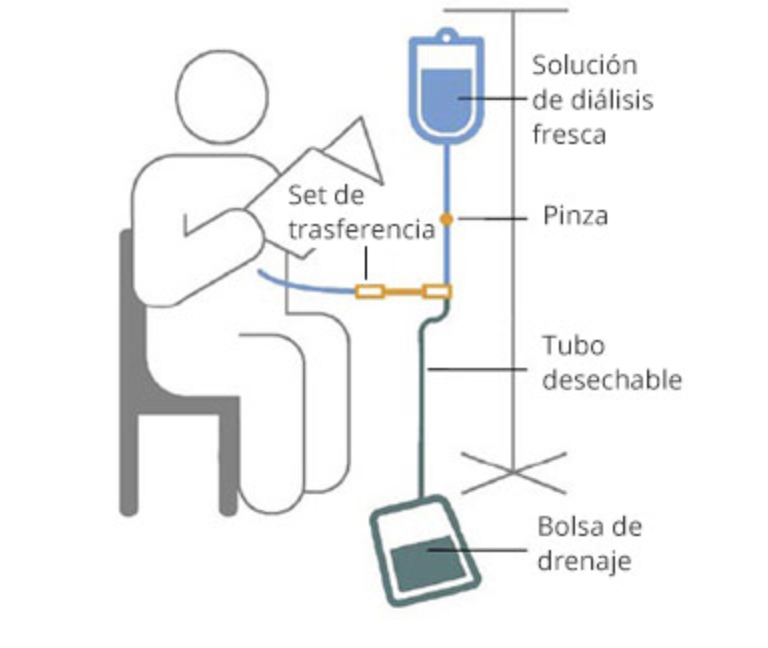
- 6 – Tratamiento médico de estados de depresión, ansiedad y angustia, deficiencias renales, sordera, ceguera o déficits neurológicos.
La Enfermedad de Von Hippel Lindau se hereda como un rasgo genético autosómico dominante, habiéndose identificado el defecto genético en el brazo corto del cromosoma 3 (3p25-26).
Fuente: https://enfermedades-raras.org/index.php/component/content/article?id=1009
Dr José Márquez Torres
MIR MAP
Dc Sergio Bea Granell
Nefrólogo CHGUV
Coordinador del Blog Renal










Comentarios recientes